Select menu: Stats | QTLs (Linkage/Association) | Data Import/Export | Load Genotypic (Marker and Map) data
Use this to import marker scores and associated map data for QTL analysis. When data are imported they can be added to the QTL data space for use within the QTL menus.
- From the menu select
Stats | QTLs (Linkage/Association) | Data Import/Export | Load Genotypic (Marker and Map) data. - Fill in the fields as required then click Save.
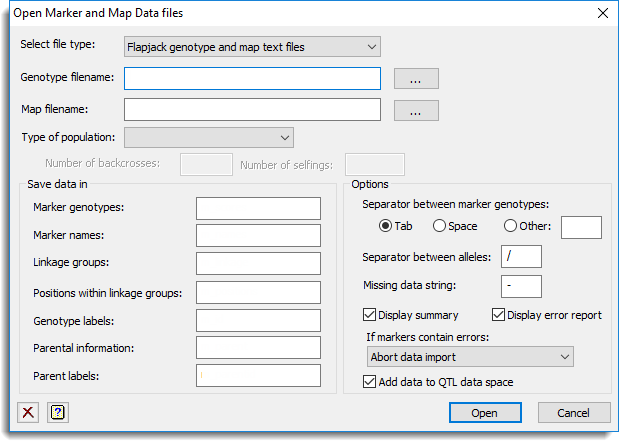
Select file type
The lists the different file types that data can be imported from. Data can be imported from Flapjack text genotype and map files (.txt), R/QTL separate genotype (csvs, csvsr) text files (.csv) and MapQTL locus genotype and map files (.loc, .map). Data from other file types can be imported using the File | Open menu item, however the structure names will not be added to the QTL data space automatically although this can be done manually using the QTL data pane.
Flapjack format
The Flapjack format contains two files:
- Genotype and map data file.
| Genotype filename | This specifies the name for the genotype data file. This file contains the information for the genotype scores for each marker. You can browse for a filename by clicking on the |
| Map filename | This file contains the map positions and linkage group information for all loci. You can browse for a filename by clicking on the |
MapQTL format
| LOC filename | This specifies the name for the locus genotype data file. This file contains the information for the type of population and the genotype scores for each locus. You can browse for a filename by clicking on the |
| Map filename | This specifies the name for the map data file. This file contains the map positions and linkage group information for all loci. You can browse for a filename by clicking on the |
R/QTL format
| CSV filename |
This specifies the marker scores, map positions and linkage groups. The .csv file format uses an extended version of the R/QTL comma-delimited separate file for genotype data (csvsr or csvs), where there is an optional column for marker sets. In the rotated .csv file the first row specifies the genotype id, and for a QTL analysis these genotype id labels should match the labels for the phenotypic genotype factor associated with the trait means. The first cell should be the name of an identifier for the genotype id, and the first row in columns 2 and 3 should be blank. Also, if marker sets are included in the file, the first row of column 4 should be left blank. Starting from row 2 in the file, the first column gives the marker names, the second column gives the linkage group for each marker, and the third column gives the positions of the markers within the linkage groups. The fourth column can be used to contain the marker sets. The remaining columns give the marker genotype scores. You can browse for a filename by clicking on the |
Type of population
A list of population types for importing from Flapjack or R/QTL format. Select as follows:
- F2 for an F2 population
- BC1 for a backcross population
- DH for a double-haploid population
- RILn for a population of recombinant inbred lines
- BCxSy for a backcross inbred lines
- CP for outcross populations
- AMP for an association mapping population.
Note that the BCxSy, CP and AMP are not supported with R/QTL files. When a population is selected the relevant fields will be enabled on the dialog.
Save data in
Specifies the data structure names to save the data in.
| Marker genotypes | Pointer | Creates a pointer containing a set of factors (one for each marker) where each factor has the same labels representing the genotype scores and the labels are all in the same order. |
| Marker names | Text | Creates a text containing the marker names. |
| Linkage groups | Factor | Creates a factor identifying the linkage group for each marker. |
| Positions within linkage groups | Variate | Creates a variate containing the positions within the linkage groups for each marker. |
| Genotype labels | Text | Creates a text containing id labels for genotypes. These can be used to ensure the genotypes for the phenotypic data matches the map data. |
| Parental information | Pointer | Creates a pointer to a set of texts (one for each parent) containing the parent information. |
| Parent labels | Text | Creates a text containing id labels for the parental information. |
Options
Provides additional options for importing the data.
| Separator between marker genotypes | Specifies the character that is used to separate the marker genotypes within a Flapjack text genotype file. You can choose either Tab, Space, or supply another character in the space provided. |
| Separator between alleles | Provides a space to specify a character to use to separate the alleles in the Flapjack text genotype # file. If no separator has been used then leave this field empty. |
| Missing data string | Specifies the character to represent missing data in a Flapjack text genotype or R/QTL file. |
| Add data to QTL data space | When selected the imported data structure names will be added to the QTL data space so that the names of the structures will be present in the QTL menus. |
Add data to QTL data space
When selected the imported data structure names will be added to the QTL data space so that the names of the structures will be present in the QTL menus.
Action Icons
| Clear | Clear all fields and list boxes. | |
| Help | Open the Help topic for this dialog. |
See also
- Importing phenotypic data for QTL analysis
- Importing genetic relationship data for QTL analysis
- Genotype and map data file format
- QTL data space for using data in QTL menus
- QTL analysis using menus