Select menu: Stats | QTLs (linkage/Association) | Data Exploration | Single Trait Linkage Analysis (Multiple Environments)
Use this to perform genome-wide scans for QTL effects (Simple and Composite Interval Mapping) in multi-environment trials. The phenotypic data required is one value for each genotype-environment combination from a set of multi-environment trials. These values will usually be predicted means obtained from a preliminary analysis of each trial. When data structures have been stored within the QTL data space then the names of these structures will be automatically entered in the relevant input fields.
- After you have imported your data, from the menu select
Stats | QTLs (linkage/Association) | QTL Analysis | Single Trait Linkage Analysis (Multiple Environments). - Fill in the fields as required then click Run.
You can set additional Options before running the analysis.
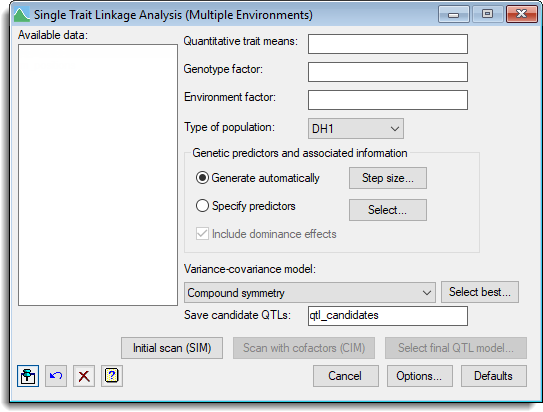
The menu is designed to provide step-by-step determination of a model to estimate QTL effects. The menu allows for the following steps:
- An initial genome-wide scan by simple interval mapping (SIM) to obtain candidate QTL positions. These can be used as cofactors in subsequent scans (composite interval mapping).
- One or more rounds of composite interval mapping (CIM), ie. a genome-wide scan for QTL effects in the presence of cofactors, which are usually potential QTL positions detected at previous steps.
- Fit a final QTL model, usually following back-selection from a set of candidate QTLS, to get a final set of estimated QTL effects.
In the case of multi-environment trials, the scans test for both QTL main effects and QTL by environment interactions.
Available data
This lists data structures appropriate to the current input field. Double-click a name to copy it to the current input field or type the name. When data has been stored in the QTL data space then by default the Available data will display only the data structures present within that data space. You can right-click on the Available data list to switch between displaying data only within the QTL data space and all data within Genstat. If the QTL data space has not been used, then all current data structures within Genstat will be displayed.
Quantitative trait means
A variate specifying the quantitative trait (phenotypic) means for each genotype by environment combination.
Genotype factor
A factor specifying the genotype for each mean.
Environment factor
A factor specifying the environment for each mean.
Type of population
A list of population types. Select as follows:
- F2 for an F2 population
- BC1 for a backcross population
- DH for a double-haploid population
- RILn for a population of recombinant inbred lines
- BCxSy for a population of backcross inbred lines, or CP for cross pollinator population.
When a population is selected the relevant fields to supply data structures will be enabled on the dialog.
Genetic predictors and associated information
By default conditional genotypic probabilities are calculated at specific linkage group positions for the QTL analysis. These options allow you to specify a step size or supply names for previously calculated genetic predictors.
| Generate automatically | When selected the genetic predictors will be generated automatically when an initial scan is run. You can click the Step size button to open a dialog where you can specify a new step size to use for the genetic predictors and associated information. If the step size is changed then the genetic predictors will be recalculated using the same names of structures specified in the QTL data space. |
| Specify predictors | Lets you specify the names of genetic predictors if they have been calculated previously. Click the Select button to open a dialog where you can specify the names of data structures for the genetic predictors. |
| Include dominance effects | For an F2 and BCxSy population select this option to include dominance effects |
Variance-covariance model
This lets you select the variance-covariance model to use across the set of environments. The Select best button opens a dialog that can be used to select the best variance-covariance matrix to model the genetic correlations between environments, based on the Schwarz (Bayes) Information Criterion (SIC or BIC) or Akaike Information Criterion (AIC). For more details about the models see the help on the Select best variance-covariance model menu.
Action buttons
| Initial scan (SIM) | Performs an initial genome-wide scan by simple interval mapping (SIM). |
| Scan with cofactors (CIM) | Opens a dialog to perform composite interval mapping (CIM) using cofactors. A default set of cofactors selected by SIM is provided and can be changed. |
| Select final model | Opens a dialog where you can establish a final model from a set of QTL candidates using backward selection and get estimates of QTL effects from this joint model. |
| Cancel | Close the dialog without further changes. |
| Options | Opens a dialog where additional options and settings can be specified. |
| Defaults | Reset options to the default settings. Clicking the right mouse on this button produces a shortcut menu where you can choose to set the options using the currently stored defaults or the Genstat default settings. |
Action Icons
| Pin | Controls whether to keep the dialog open when you click Run. When the pin is down |
|
| Restore | Restore names into edit fields and default settings. | |
| Clear | Clear all fields and list boxes. | |
| Help | Open the Help topic for this dialog. |
See also
- QTL data space for using data in QTL menus
- QTL analysis using menus
- Linkage analysis options dialog.
- Multi-trait linkage analysis (single environment) menu
- Single trait linkage analysis (single environment) menu.
- Specifying names of genetic predictors.
- Calculate genetic predictors menu.
- Select best variance-covariance model menu.
- QMQTLSCAN procedure for QTL genome scans in multi-environments in command mode.
- QCANDIDATES procedure for selecting QTLs in command mode.